Case Summary
A 38 year old female came with:
- Headache for 6 months
- Diminution of peripheral vision bilatetrally
- Irregular menstrual cycle for the last 1 year
- Now, polydypsia and excessive urination
Her prolactin level was 115.6 ng/ml (increased), FSH was 1.3 mIU/ml (decreased) and LH was 0.242 mIU/ml (decreased).
CT scan of head showed moderately enhancing hypodense lesion measuring approximately 3.5 X 3 cm seen in suprasellar region suggestive of pituitary tumor.
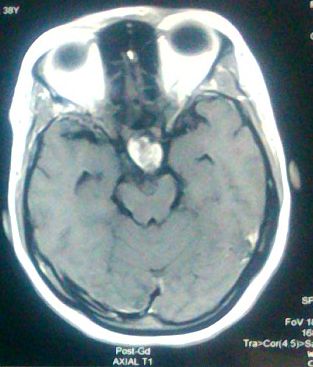
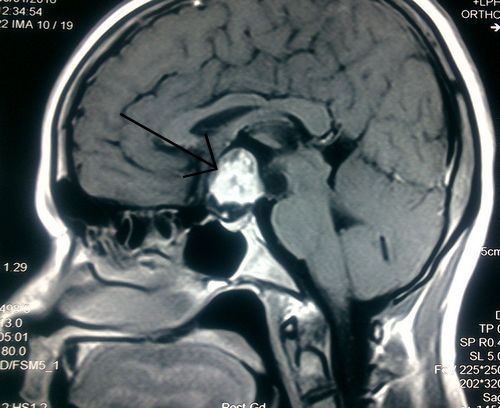
MRI brain showed mixed signal intensity suprasellar lesion ~ 3.18 X 2.19 X 3.09 cm showing intense enhancement of solid component and rim enhancement of cystic component suggestive of Craniopharyngiona. Portion of the lesion is projecting into sella causing compression and displacement of pituitary gland posteromedially. Cystic component of the lesion is slightly compressing 3rd ventricle and displacing it towards left side.
Visual perimetry showed bitemporal hemianopia.

Craniotomy and complete excision was done.
Case discussion in short
The two common sellar and parasellar tumors are pituitary adenoma and craniopharyngioma.
Similarities:
- Both are histologically benign and slow-growing
- Both have tendency to cause similar symptoms like progressive visual loss, progressive endocrine problems and headache.
- Both have an excellent prognosis if treated suitably with surgical management and occasionaly with adjunctive medical treatment or radiotherapeutic procedures including radiosurgery.
Differences:
- Pituitary adenomas are more common (3rd most common intracranial tumor and about 80% of sellar tumors) while, Cranipharyngiomas comprise only 1-3% of intracranial tumors.
- Pituitary adenomas affect mainly adults while, the incidence of cranopharyngioma is bimodal (peak incidence in children 5-14 years and older adults 65-74 years)
- Pituitary adenomas originate from the cells of anterior lobe of pituitary gland, while Craniopharyngiomas are believed to originate from ectoblastic remnants of Rathke’s pouch.
Craniopharyngioma
Thought to arise from embryological remnants of the craniopharyngeal duct, these benign non-glial epithelial neoplasms with solid, cystic, and calcified components can arise anywhere along an axis from the third ventricle to the pituitary gland. Around 95% of these have a suprasellar component (20-40% are purely suprasellar, 50-75% have both supra- and intra-sellar components and only about 5% are purely intrasellar). Occasionally, a suprasellar tumor extends into the anterior (9%), middle (8%), or posterior (12%) fossa. Other rare locations include the nasopharynx, paranasal area, sphenoid bone, ethmoid sinus, intrachiasmatic area, temporal lobe, pineal gland, posterior cranial fossa, cerebellopontine angle, midportion of the midbrain, or completely within the third ventricle. Common findings include headache, vision loss, diabetes insipidus, panhypopituitarism, short stature, hypothalamic dysfunction with behavioral and memory disturbances, hyperphagia, and obesity. The unfavorable prognostic factors are age of patient under 5 years, tumor size >5 cm and hydrocephalus.
3 major clinical syndromes based on location of craniopharyngiomas:
- Prechiasmal/Chiasmal: Compression of optic apparatus – optic atrophy, bitemporal hemianopia
- Retrochiasmal: 3rd ventricle obstruction – hydrocephalus with signs of raised ICP
- Intrasellar: Compression of pituitary stalk and hypothalamic region – Endocrinopathy and headance
H&E stained section from Craniopharyngioma demonstrates:
- Adamantinomatous pattern (hard): with a basal layer of columnar cells separated by loosely arranged stellate cells. Palisading epithelial cells with keratinization and calcification are prominent.
- Supports embryonic origin: Arises from neoplastic transformation of embryonic squamous cell nests of the involuted craniopharyngeal duct that initially connects Rathke’s pouch to the stomodeum.
- Papillary variant: is more often seen in adults and contains papillae of well-differentiated squamous epithelium (no calcification).
- Supports metaplastic origin: Arises from metaplasia of the adenohypophyseal cells in the pars tuberalis of the adenohypophysis, resulting in the formation of squamous cell nests.
Radiologically, calcifications are most readily detected by computed tomography. When imaged by magnetic resonance scanning, craniopharyngioma has a solid component as well as a cystic or a multicystic component with intermediate fluid density.
Treatment philosophy:
If the tumor is favorably localized, the therapy of choice is complete resection, with care taken to preserve optical and hypothalamic functions. In patients with unfavorable tumor localization (ie, hypothalamic involvement), recommended therapy is a limited hypothalamus-sparing surgical strategy followed by local irradiation.
Explanation of the clinical presentation in the case above
A. Endocrine manifestations:
Endocrine manifestations are the result of compression of pituitary stalk or pituitary gland resulting in a state of hypopituitarism.
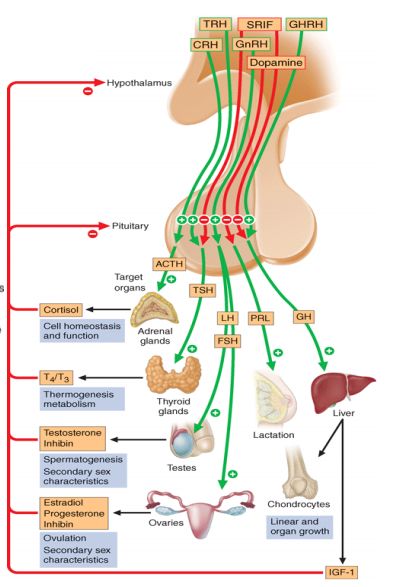
- High prolactin level:
- Elevated prolactin levels are expected findings in cases of functional pituitary adenoma i.e. prolactinoma. But in cases of other pituitary adenomas and craniopharyngiomas, elevated prolactin level is explained by the “stalk effect”. This may be seen with any disease within or near the pituitary gland and stalk that interferes with the delivery of dopamine (a neurotransmitter – inhibitory to prolactin secreting cells of pituitary) from the hypothalamus to the prolactin secreting cells of the pituitary. Elevated prolactin levels manifest as menstrual abnormalities, subfertility and galactorrhea.
- LH/FSH deficiency: Decreased gonadotropin levels in such cases can be explained by 2 mechanisms –
- Hypopituitarism resulting from decreased delivery of GnRH to pituitary gland
- Elevated prolactin levels: The reason hyperprolactinemia causes infertility, is the result of its inhibitory effect on the release of GnRH from hypothalamus. This results in decreased release of gonadotropins (LH and FSH) from the pituitary gland.
- Irregular menstruation: The anterior lobe of pituitary secretes gonadotropins in response to GnRH from the hypothalamus to produce an effect on the ovary. Follicular development is driven by FSH, but lutenizing hormone (LH) is responsible for ovulation. FSH acts on theca cells to induce LH receptor expression and render the cells sensitive to LH. LH also stimulates the theca cells to produce androstenedione, which is converted to estradiol by granulosa cells. The estradiol produced further stimulates LH release from the pituitary. When a critical level of LH is reached, ovulation occurs and the follicle rapidly changes to a corpus luteum. Progesterone, produced by the corpus luteum, increases following ovulation and inhibits LH secretion by an effect on the hypothalamus.Menstrual irregularities and amenorrhea is due to anovulation resulting from: decreased gonadotropins as explained above. Stimulation with GnRH does not produce an increase in gonadotropins suggesting a dysfunction in the blood supply to the pituitary due to the tumor.
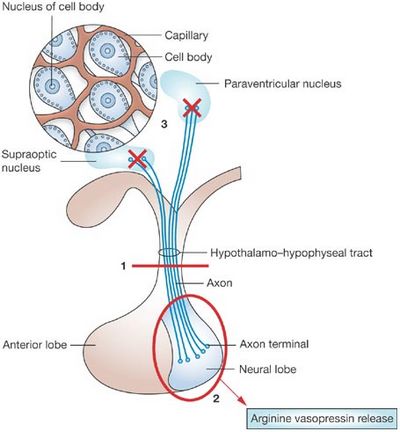
- Polyuria and Polydipsia: These are the manifestations of Diabetes Insipidus (DI) resulting from Vasopressin (ADH) insufficiency. ADH or AVP (arginine vasopressin) is produced by magnocellular cells in Paraventricular nucleus (PVN) of hypothalamus and Supra-optic nucleus (SON) which is transported and stored in posterior lobe of pituitary gland within neurosecretory granules through the infundibulum/pituitary stalk. ADH also has important local action when transported to the anterior pituitary lobe where it stimulates corticotrophic cells synergistically with CRH to produce ACTH. Craniopharyngioma results in central DI which can be attributed to:
- Hypothalamo-pituitary axis dysfunction: resulting from compression of pituitary stalk
- Damage of hypothalamus
Central diabetes insipidus (DI) is known to occur preoperatively in 8-35%, and postoperatively in 70-90% of patients with a craniopharyngioma. Extensive damage to magnocellular cells of the hypothalamus (supraoptic and paraventricular nuclei) leads to permanent DI, especially if the pituitary stalk section is in proximity to or directly involves the median eminence. The classical triphasic response starts with a transient DI due to AVP deficiency, followed by the syndrome of inappropriate antidiuretic secretion (SIADH) due to the release of AVP from disintegrating magnocellular cells, finally leading to permanent DI over a few weeks.
Other features of stalk compression are: Growth hormone deficiency, ACTH deficiency, TSH deficiency. Hypothalamic involvement can lead to weight gain/hyperphagia and personality changes.
B. Visual field defects:
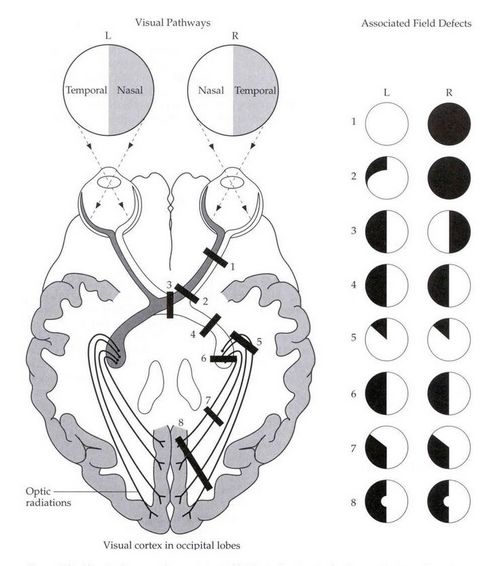
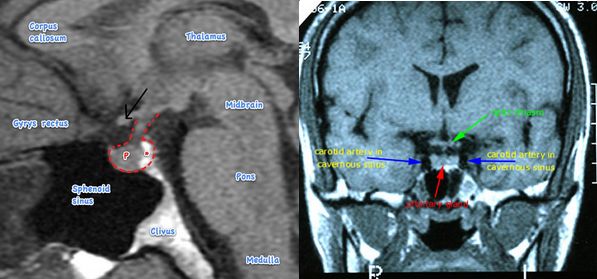
The patient had bitemporal hemianopia which is a diagnostic feature of a lesion in the optic chiasma. In the visual pathway, the two optic nerves join at the optic chiasma, where the nasal fibers responsible for the temporal visual field, decussate. Hence, a lesion in the optic chiasma affects temporal vision in both the eyes. Sella turcica/hypophyseal fossa sits on a depression in the body of the sphenoid bone and contains the pituitary gland/hypophysis. It is located behind the chiasmatic groove and tiberculum sellae. The suprasellar component of craniopharyngioma can compress the optic chiasm, which lies on top of the pituitary, enveloping the pituitary stalk.
Another cause of diminution of vision in craniopharyngioma is due to hydrocephalus leading to papilledema, which was not present in our case.
C. Headache:
Headache in craniopharyngioma occurs by 3 mechanisms:
- Raised ICP resulting from tumor mass itself
- Raised ICP resulting from compression of 3rd ventricle leading to obstructive hydrocephalus
- Meningeal irritation due to leaked out cyst
Since, there was hydrocephalus and no signs of meningeal irritation, the probable cause of headache in this patient was due to raised ICP resulting from the tumor mass itself.

He is the section editor of Orthopedics in Epomedicine. He searches for and share simpler ways to make complicated medical topics simple. He also loves writing poetry, listening and playing music. He is currently pursuing Fellowship in Hip, Pelvi-acetabulum and Arthroplasty at B&B Hospital.