Fatty Acid Activation (Cytoplasm)
Before undergoing oxidation, fatty acid must be activated first by conversion into Acyl-CoA by the action of Fatty Acyl-CoA synthase (thiokinase).
- ATP is converted to AMP (not ADP) – 2 ATP equivalent used
- Uses Mg2+ as cofactor, Co-ASH and ATP
Sites of Beta-oxidation
A. Mitochondria:
- Short/Medium chain FA (2-12 C): Diffuse directly into mitochondria
- Long chain FA or LCFA (12-20 C): Uses carnitine shuttle to enter mitochondria (inner mitochondrial membrane is impermeable to FA)
B. Perioxisomes:
- Very long chain FA or VLCFA (>20 C)
- After shortening of VLCFA in perioxisome, it is transferred to mitochondria for further oxidation.
- Perioxismoes are also the site of alpha-oxidation (FA with methyl-group at beta-carbon which block beta-oxidation).
Omega oxidation (occurs in endoplasmic reticulum) becomes important when beta-oxidation is impaired.
Zellweger’s (Cerebrohepatorenal) syndrome: Absence of perioxisome in all tissues (includes neonatal adrenoleukodystrophy and infantile refsum’s disease).
- Accumulation of VLCFA
- Accumulation in brain leads to severe neurological symptoms
X-linked adrenoleukodystrophy: Inability to transport VLCFA across perioxismal membrane.
- Accumulation of VLCFA
Refsum’s disease: Defect in alpha-oxidation of phytanic acid
- Normal VLCFA, with accumulation of phytanic acid (found in dairy products and ruminant fat and meat)
- Neurological symptoms
It occurs in hepatocytes, myocytes, adipocytes.
- Neurons cannot use FA as energy (do not cross BBB)
- RBC cannot use FA as energy (no mitochondria)
Carnitine Shuttle
Carnitine (produced from lysine in liver and kidney cells) is added to activated FA (acyl-CoA) in the intermembrane space of the mitochondria by Carnitine Palmitoyl Transferase-1 (CPT-1).
- CPT-1 is inhibited by malonyl-CoA so as to prevent newly synthesized FAs from being degraded.
- Problem in lysine metabolism leads to disturbance in formation of carnitine and consequently FA metabolism and storage of TGs in cells.
CPT-1 deficiency:
- Affects liver
- Inability to use fatty acid (also for ketogenesis) during fasting leading to non-ketotic hypoglycemia
CPT-1 inhibition: Malonyl-CoA and Sulfonylureas (anti-diabetic drugs)
Acyl-Carnitine is transported into the matrix by crossing the innter mitochondrial membrane by the carnitine transporter (Carnitine Acyl carnintine translocase).
In the mitochondrial matrix, carnitine is exchanged for CoA by CPT-2 and acyl-carnitine is changed back to acyl-CoA (activated FA).
CPT-2 deficiency:
- Affects cardiac and skeletal muscles
- Cardiomyopathy, myoglobinemia and weakness after exercise
- ↑ TG content in muscles (unable to use as energy)
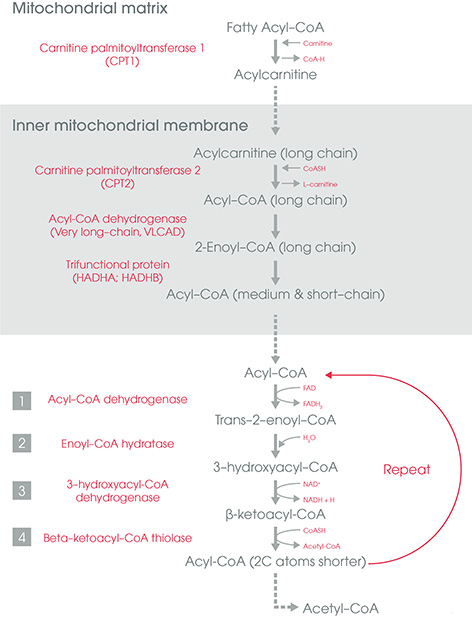
Beta-Oxidation (Mitochondrial Cytosol)
It is the reversal of FA synthesis. 1st step is catalyzed by one of a family of four acyl-CoA dehydrogenases that each has a specificity for either SCFA, MCFA, LCFA or VLCFA.
Inhibition of Short Chain and Medium Chain Fatty Acyl CoA Dehydrogenase (SCAD and MCAD): Hypoglycin toxin in Ackee fruit (Jamaican vomiting sickness)
MCAD deficiency:
- autosomal recessive
- non-ketotic hypoglycemia (liver unable to produce enough energy from beta-oxidation to supply gluconeogenesis and ketones from β-oxidation)
- C6-C10 acyl carnitines in the blood (liver unable to break FAs down further than C8-C10) and C6-C10 dicarbocylic aciduria
- symptoms often precipitated by infection or stress
- treatment: low fat diet with frequent meals of high carbs
At the end of 4 reactions (oxidation, hydration, oxidation and reverse condensation) –
- Product: (n-2) C Acyl-CoA i.e. Acyl-CoA with 2 C less than precursor + Acetyl-CoA
- Along with acetyl-CoA, NADH and FADH2 is also produced.
- Reaction repeats until FA is completely degraded to Acetyl-CoA (last reaction produces 2 acetyl-CoA) and Propionyl-CoA in case of odd chain fatty acid (propionyl CoA is converted to methymalonyl-CoA by a reaction requiring biotin and then to succinyl-CoA by methylmalonyl-CoA mutase requiring vitamin B12)
Genetic error in methylmalonyl-CoA mutase or vitamin B12 deficiency causes methylmalonyl acidemia and aciduria.
Energy balance in Fatty acid oxidation
- 1 NADH = 2.5 ATP (3 in old concept)
- 1 FADH2 = 1.5 ATP (2 in old concept)
- 1 GTP = 1 ATP
- 1 Acetyl-CoA = 10-12 ATP (3 NADH + 1 FADH2 + 1 GTP)
- 1 Propionyl-CoA (Succinyl-CoA) = 5-6 ATP (1 NADH + 1 FADH2 + 1 GTP)
- 1 cycle of beta-oxidation = 4-5 ATP (1 FADH2 + 1 NADH)
- ATP spent in fatty acid activation = 2 ATP
- Number of acetyl-CoA generated = number of carbon in precursor fatty acid/2
- Number of beta-oxidation cycles = Number of acetyl-CoA generated – 1
For odd chain fatty acids:
- 1 propionyl-CoA (3C) is generated at the end
- Number of acetyl-CoA generated = (number of carbon in precursor fatty acid – 3)/2
- Number of beta-oxidation cycles = Number of acetyl-CoA generated
Number of carbon in precursor = Number of carbon in products
- 16 C = 8 X 2 C (acetyl-CoA)
- 17 C = 3 C (propionyl-CoA) + 7 X 2 C (acetyl-CoA)
Example:
1. Palmitic acid (16 C):
- Generates 8 Acetyl-CoA + Undergoes 7 cycles of beta-oxidation – 2 ATP for FA activation
- 80 to 96 ATP + 28 to 35 ATP – 2 ATP = 106 ATP (new concept) to 129 ATP (old concept)
2. 17 C fatty acid:
- Generates 1 Propionyl-CoA + Generates 7 Acetyl-CoA + Undergoes 7 cycle of beta-oxidation – 2 ATP for FA activation
- 5 to 6 ATP + 70 to 84 ATP + 28 to 35 ATP – 2 ATP = 101 (new concept) to 123 ATP (old concept)