Purines and Pyrimidines Bases
Purines = 2 rings
- Adenine
- Guanine
- Hypoxanthine (Deaminated Adenine)
- Adenine to Hypoxanthine deamination is mediated by Adenosine deaminase which is decreased in Autosomal recessive SCID. Accumulated dATP inhibit ribonucleotide reductase leading to deficient synthesis of other deoxyribonulceotide precursors for DNA synthesis.
- Xanthine (Deaminated Guanine)
Mnemonics:
Pure As Gold i.e. Purines are Adenine and Guanine.
How to identify purines?
1. First look for 2 rings. If 2 rings are present, then –
2. Look for location of NH2- (amino) group:
- 12 o’ clock (A is the 1st letter): Adenine
- 7 o’ clock (G is the 7th letter): Guanine
- Non NH2- group: Hypoxanthine or Xanthine
3. If there is no NH2- group, look of O- group:
- 1 O- group: Hypoxanthine (hypo means less)
- 2 O- group: Xanthine
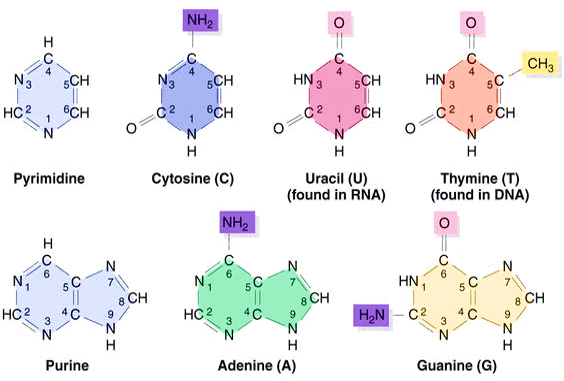
Pyrimidines = 1 ring
- Cytosine
- Uracil (Deaminated Cytosine) – used to identify RNA (Northern blot)
- Thymine (Methylated Uracil)
- Thymine requires 2 “T”s – Thymylate synthase and Tetrahydrofolate (THF).
- Uracil to thymidine methylation is mediated by Thymidylate synthase which is inhibited by 5-FU. This reaction requires Tetrahydrofolate (THF) which is synthesized from Dihydrofolate (DHF) by DHF reductase i.e. inhibited by Methotrexate (eukaryotic), Trimethoprim (prokaryotic) and Pyrimethamine (protozoal).
- used to identify DNA (Southern blot)
Mnemonics:
Pyrimidines are CUT from Purines: Cytosine, Uracil and Thymine
How to identify pyrimidines?
1. Look for a single ring. If single ring is present:
2. Remember in sequence of CUT:
- Cytosine has NH2- group.
- Uracil in RNA (Deaminated cytosine) has no NH2- group.
- Thymine in DNA (Methylated uracil) has CH3- group.
Complementary Purines and Pyrimidines
Adenine (A) pairs via 2 hydrogen bonds to Uracil (U) in RNA or Thymine (T) in DNA, i.e. A=U or T.
Guanine (G) pairs via 3 hydrogen bonds to Cytosine (C), i.e. G ≡ C.
Chargaff’s rule:
- % purines = % pyrimidines
- % A = % T (%U)
- $G = % C
Nucleosides, Nucleotides and Nucleic acids
Nucleosides = Base + Pentose sugar (Ribose for RNA and Deoxyribose for DNA) e.g. (deoxy-)adenosine, (deoxy-)guanosine, (deoxy)-cytidine, (deoxy)-uridine, deoxythimidine
Nucleotides = Base + Pentose sugar + Phosphate group e.g. AMP, ADP, ATP, dAMP, dADP, dATP.
Ribonucleotide reductase: forms deoxyribonucleotides from ribonucleotides.
- Inhibited by Hydroxyurea.
- Inhibited by dATP (accumulated in ADA SCID).
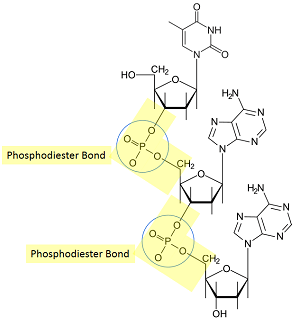
Nucleic acid = Nucleotides linked by 3′,5′ phosphodiester bonds.
- Phosphate group at 5′ position is connected to 3′ position of another nucleotide.
- Base sequence is written in 5’→3′ direction (left to right) e.g. 5′-TCAG-3′ or TCAG.
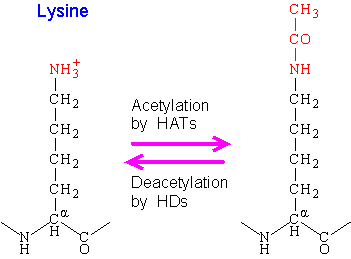
Histones
- Histones are rich in: Arginine and Lysine, hence POSITIVE charged.
- DNA have PO4³-, hence NEGATIVELY charged.
- Histones (positive) is attracted to DNA (negative) and DNA is condensed.
- Histone Acetylation by Histone Acetyltransferase (HAT) removes positive charge (from lysine) – Histones are repelled from DNA and DNA is relaxed and genomes are available for transcription, i.e. transcriptionally active: Euchromatin
- Inhibited in Huntington’s disease.
- Histone Deacetylation by Histone Deacetylase (HDA) reverses acetylation of histone – Histones condeses the DNA and genomes are not available for transcription, i.e. transcriptionally inactive: Heterchromatin.
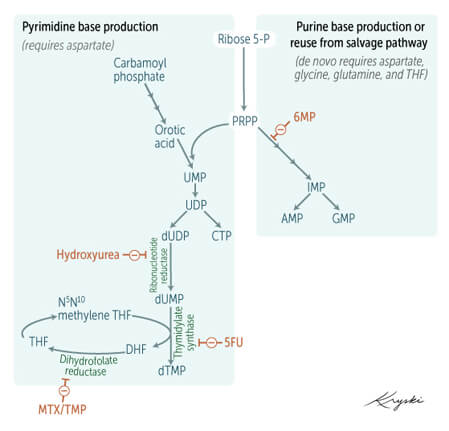
Purine and Pyrimidine De-novo synthesis (Occurs in Liver)
Nucleotide synthesis sequence:
- Sugar = Ribose-5-Phosphate (from pentose phosphate pathway)
- Sugar + Phosphate (from ATP) = Ribose-5-Phosphate + ATP (PRPP synthase) = PRPP (Phosphoribosyl pyrophosphate)
- Rate-limiting step inhibited by nucleotides
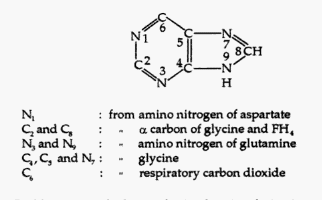
- (Sugar + Phosphate) i.e. PRPP + Nitrogen base = Mono-nucleotide (OMP for pyrimidine and IMP for purine)
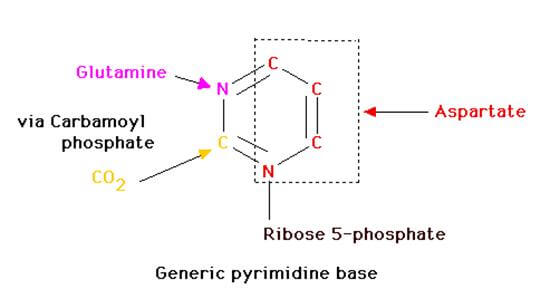
- Nitrogen base for pyrimidine = Orotic acid (Glutamine and aspartate)
- Orotate phosphoribosyl transferase (Phosphate and sugar added to nitrogen base)
- Nitrogen base for purine = Glutamine and Glycine
- Phosphoribosyl amidotransferase (Nitrogen base added to phosphate and sugar)
- Inhibited by negative feedback from purine nucleotides and analogues likes:
- AMP, IMP, GMP
- Allopurinol
- 6-Mercaptopurine and Azathioprine (which releases 6-Mercaptopurine).
- Nitrogen base for pyrimidine = Orotic acid (Glutamine and aspartate)
- Synthesis of UMP (pydrimidine) and IMP (purine):
- OMP —(OMP decarboxylase) —> UMP
- IMP (use ATP) = XMP (xanthine monophosphate) –> GMP
- IMP (use GTP) = AMP
- Ribonucleotide (NDP) —(ribonucleotide reductase)—> deoxy-ribonucleotide (dNDP)
- Inhibited by Hydroxyurea
- Deaminated uracil = Cytosine (UTP to CTP)
- Methylated uracil = Thymine (dUMP to dTMP)
- Requires thymidylate synthase requiring folate (methylene-THF as CH3 donor) and vitamin B12
- Thymidylate synthase is inhibted by 5-FU.
- DHFR converting DHF to THF is inhibited by Methotrexate (eukaryocytes) and Trimethoprim (prokaryotes).
Orotic aciduria: defect in UMP synthesis pathway. Deficient in orotate phosphoribosyl transferase/OPRT (converts orotate to OMP), or in OMP decarboxylase (converts orotidine-5-monophosphate to UMP).
- Type I orotic aciduria = both OPRT and OMP decarboxylase
- Type II orotic aciduria = OMP decarboxylase
Lead to build up of orotate, which is found in urine. Causes retarted growth and severe megalobasltic anemia (DNA synthesis block) without hyperammonemia (no urea cycle block).
Treat by administering uridine and/or cytidine. They get phosphorylated to for their respective NMP, NDP, and NTPs, which lead to feedback inhibition of pyrimidine biosynthesis pathway, thus preventing orotic acid synthesis (inhibit first step: carbamoyl phosphate synthetase II).
Orotic acid synthesis for Pyrimidine synthesis:
- CO2 + Glutamine —CPS2—> Carbamoyl phosphate (CAP)
- CAP + Aspartate —aspartate transcarbamoylase—> Carbamoyl aspartate (CAA)
- rate-limiting step (inhibited by CTP and UTP and inhibition reversedby ATP)
- CAA —dihydroorotase & dihydroorotase dehydrogenase —> Orotic acid
Carbamoyl Phosphate Synthetase-2 of Pyrimidine synthesis is present in cytoplasm, unlike CPS-1 of urea cycle which is mitochondrial. When carbamoyl phosphate accumulates in mitochondria (urea cycle) it can leak out into cytoplasm and participate in pyrimidine synthesis (Ornithine Transcarbamoylase or OTC deficiency). Hence, accumulation of orotic acid and orotic aciduria is seen in:
Pyrimidine synthesis:
Purine synthesis:
Purine Metabolism (Occurs outside Liver)
First: Phosphate is lost from nucleotide and nucleoside formed (Nucleotide – Phosphate = Nucleoside)
Second: Sugar is lost from nucleoside and purine base is left (Nucleoside – Sugar = Purine base)
Third: Purine bases now have 2 options –
- Recycle through Salvage pathway with HGPRT (Hypoxanthine Guanyl Phosphoribosyl Transferase) that adds ribose-phosphate to purine base to form nucleotide again (IMP and GMP) – 90%.
- Near-complete deficiency of HGPRT activity is seen in Lesch-Nyhan syndrome (X-linked recessive). Cells of basal ganglia normally have very high HGPRT activity.
- Lesch-Nyhan syndrome presents with: Spastic cerebral palsy (basal ganglia involvement), mental retardation, self-mutilation of hands and lips, hyperuricemia (salvage pathway blocked leading to increased share of excretion pathway in which uric acid is formed) and early death.
- Early sign of Lesch-Nyhan syndrome: Orange urine crystals in diaper
- Excreted in urine as uric acid mediated by Xanthine Oxidase – 10%.
- Xanthine oxidase inhibitors: Purine analogue (Allopurinol – also inhibits PRPP amidotransferase in purine synthesis) and Non-purine analogue (Febuxostat – lesser adverse effects).
- Lactate and urate compete for the same transport system in kidney: Hence, conditions leading to lactic acidosis cause hyperuricemia (Alcoholism, Von-Gierke’s disease i.e. Glucose 6-phosphatase deficiency).
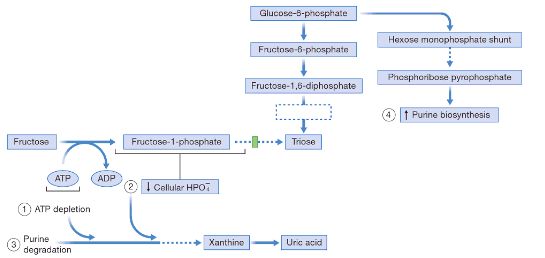
Phosphorylated sugar accumulation cause hyperuricemia:
1. Glucose-6-Phosphate (in Von-Gierke’s disease or Glucose 6-Phosphatase deficiency): Glucose 6-Phosphate accumulates which undergoes HMP shunt to generate excessive ribose-5-phosphate, the precursor of purines.
2. Phosphorylated sugars (Fructose 1-phosphate in Aldolase B deficiency i.e. hereditary fructose intolerance and Galactose 1-phosphate in G1PUT deficiency i.e. Classic galactosemia) accumulation leads to ATP depletion and Pi sequestration in sugar. Nucleotides and intracellular Pi must be in balance. When Pi decreases, liver AMP deaminase is activated leading to degradation of AMP into uric acid leading to hyperuricemia.
preparing for step exams and this is by far the most important website for preparing biochemistry. Not only basics are covered, multiple topics are interlinked which helps to solve step 1 questions. i wish more such biochemistry topics are covered.