Propionyl CoA is a common intermediate in catabolism of essential amino acids and odd chain fatty acids. It is also called VOMIT pathway which stands for:
- Valine
- Odd chain fatty acids
- Methionine
- Isoleucine
- Threonine
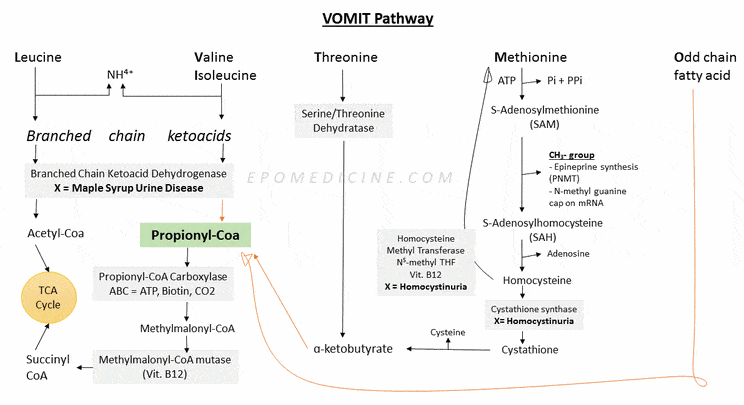
They enter TCA cycle – using PMS pathway:
- Propionyl CoA
- Methylmalonyl CoA
- Succinyl CoA
Defects in VOMIT Pathway
Defect in Branched Chain Ketoacid Dehydrogenase (BCKD) – Maple Syrup Urine Disease (MSUD)
Defect in enzyme Branched Chain Ketoacid Dehydrogenase:
- Enzyme similar to pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase – requires thiamine, lipoic acid, CoA, FAD, NAD+
- Defect in BCKD leads to elevation of plasma concentrations of the branched-chain amino acids and corresponding keto acids
- Present in Inner mitochondrial membrane
Features:
- Infantile onset – normal for first week; progressive symptoms
- Leucine accumulation – Neurological symptoms
- Impair regulation of cell volume – resulting in decreased serum sodium concentration and increased intracellular water, leading to cerebral edema.
- Another mechanism of neurotoxicity may be increased production of glutamate, glutamine, and gamma-aminobutyric acid (GABA) caused by the rapid transport of leucine across the blood-brain barrier.
- Features: Mental retardation, Abnormal muscle tone
- Isoleucine accumulation – Maple syrup odor of urine
- Caramel colored urine
- Ketosis, Coma and Death
Treatment: Restriction of dietary valine, leucine and isoleucine
Defect in Propionyl-CoA Carboxylase and Methylmalonyl-CoA Mutase
Inability to handle all 5 components of VOMIT
Propionyl-CoA carboxylase requires Biotin (Vit.B7) and Methylmalonyl-CoA Mutase requires Cobalamin (Vit. B12)
Pathophysiology:
- Accumulated organic acids inhibit gluconeogenesis: hypoglycemia
- Fatty acids are transported into mitochondria as carnitine conjugates where they are b-oxidized to ketones: ketosis.
- Organic acid inhibits the urea cycle: hyperammonemia
- Organic acid inhibits glycine degradation: hyperglycinemia
- Organic acid inhibits hematopoeisis: neutropenia
Features:
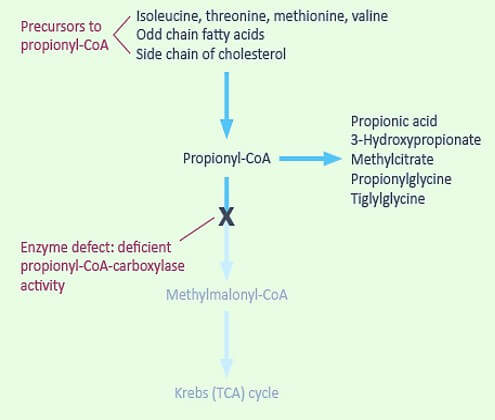
- Ketoacidosis – due to failure to metabolize ketoacids produced from 4 amino acids
- Propionyl-CoA carboxylase deficiency (Propionic acidemia): Accumulation of –
- Propionic acid
- Methyl citrate
- Hydroxypropionic acid
- Methylmalonyl-CoA mutase deficiency: Accumulation of-
- Methylmalnate leading to Methylmalonic aciduria
Treatment: Diet low in valine, methionine, isoleucine and threonine
Homocystinemia/Homocystinuria
Causes:
- Cystathione synthase deficiency (requires vitamin B6)
- Decreased affinity of cystathione synthase for Pyridoxal Phosphate (B6)
- Homocysteine methyl transferase deficiency (requires vitamin B12)
- Deficiency of Folate, Vitamin B6 or B12
Defect:
- Inability to re-methylate homocysteine to methionine (Vit B12, folate and homocysteine methyl transferase deficiency)
- Inability to make cystathione to go into propionic acid pathway (Cystathione synthase and Vit. B6 deficiency)
Pathophysiology:
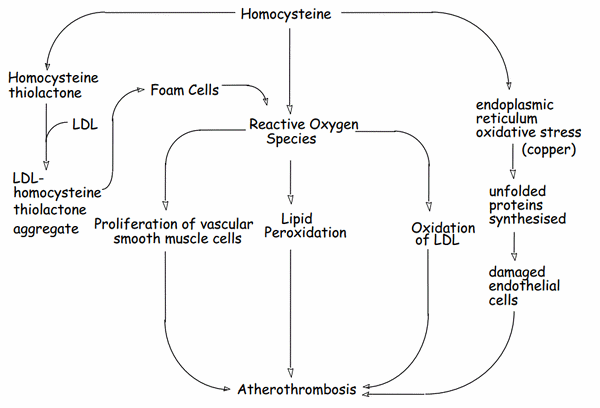
Homocysteines have “-SH” groups which are reducing agents.
Atherosclerosis:
Marfan-like syndrome:
- Fibrillin-1 contains ∼13% cysteine residues and can be modified by homocysteine.
Presentation:
- DVT, Stroke, Myocardial infarction
- Marfan-like: Mental retardation, Lens dislocation (downward as opposed to Marfan’s syndrome in which dislocation is outward and upward), tall habitus with long extremities