Although, we classify as intravascular and extravascular hemolysis, “diseases” don’t read the book. These disorders may be described as causing extravascular hemolysis, but your case may be the uncommon exception with intravascular hemolysis that was not mentioned. Diseases may cause anemia by both intravascular and extravascular hemolysis. Extravascular hemolysis typically accompanies intravascular hemolysis. Disorders may also switch from one to another.
Intravascular hemolysis vs Extravascular hemolysis
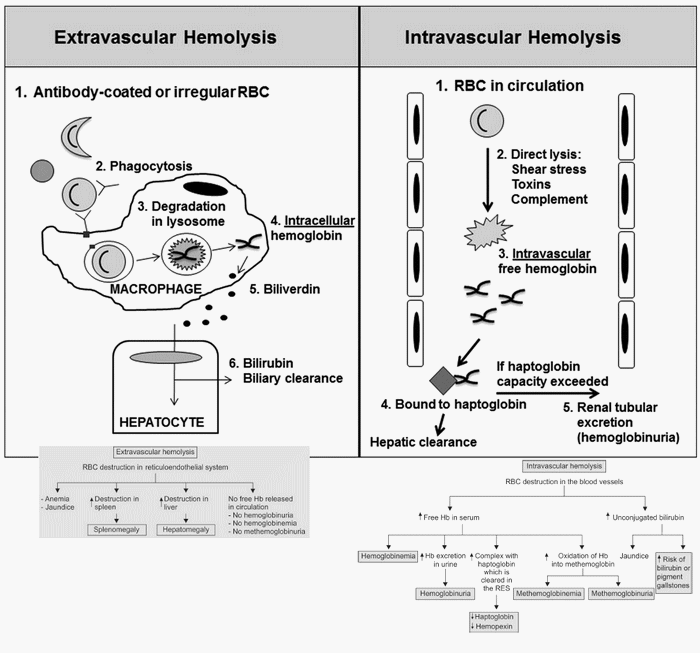
| Intravascular hemolysis | Extravascular hemolysis | |
| Site of hemolysis | Within the circulation (vasculature) | Mononuclear phagocytes of Eeticuloendothelial system i.e. spleen and liver macrophages |
| Free hemoglobin | Released in blood | Engulfed by macrophages |
| Fate of free hemoglobin | Binds to haptoglobin
|
Converted to bilirubin |
| Serum haptoglobin | Decreased or absent | Normal or slightly decreased |
| What happens when haptoglobin is depleted | Free heme binds to hemopexin or Hemoglobin is oxidized to Methemoglobin. | |
| Serum hemopexin | Decreased | Normal |
| In blood | Hemoglobinemia
Methemoglobinemia Mild unconjugated hyperbilirubinemia Increased LDH |
Moderate unconjugated hyperbilirubinemia
|
| Consequences of hemoglobinemia (nitric oxide scavenging capacity of hemoglobin) | Renal failure
Hypertension Smooth muscle spasm Prothrombotic state |
No hemoglobinemia |
| In urine | Hemoglobinuria (brown)
Hemosiderinuria |
Increased urobilinogen and urobilin (yellow) |
| Spleen and Liver | Normal | Enlarged (Work hypertrophy) |
| Kidneys | Iron and hemosiderin deposition leading to AKI | Normal |
| Peripheral blood smear | Schistocytes/Burr cells/Helmet cells/Triangle cells (Microangiopathic hemolytic anemia like DIC, malignant hypertension, SLE, TTP, HUS)
Heinz body (G6PD deficiency) – induce cell membrane damage
|
Spherocytes (Spherocytosis, AIHA, G6PD deficiency)
Spherocytosis not corrected by addition of glucose; High MCHC (Hereditary spherocytosis) Bite cells (G6PD deficiency) Target cells, Howel-Jolly bodies (Sickle cell disease, Beta thalassemia major) Sickle cell, Polychromatophilia (Sickle cell disease) Basophilic stipplings (Beta thalassemia major) |
| Direct Antiglobulin or Direct Coomb’s Test (DAT/DCT) | Positive in Cold Hemolysin AIHA (antibodies react at 4-6⁰c and dissociate at ≥3o ⁰c) | Positive in Cold Agglutinin AIHA (antibodies react at 4-6⁰c and dissociate at ≥3o ⁰c)
Warm AIHA (≥37 ⁰c) |
| IgG against P blood group antigen (Donath-Landsteiner antibody) | IgM (Cold AIHA) and IgG (Warm AIHA) | |
| Paraoxysmal Cold Hemoglobinuria | Warm AIHA – Primary (idiopathic), SLE, RA, B cell lymphoid neoplasms, Drugs (methyldopa, penicillin)
Cold agglutinin AIHA – Mycoplasma, Infectious mononucleosis |
|
| Osmotic fragility test (Pink test) | Negative | Positive in Hereditary spherocytosis |
| Sickling test (2% metabisulfite or dithionite) | Negative | Sickling in Sickle cell anemia. |
| NESTROF Test (0.35% NS added to control and patient’s blood sample) | Screening test for Beta-thalassemia trait – black line in white paper placed behind the patient’s tube is not visible (resistant to hemolysis). | |
| Hb Electrophoresis | Sickle cell trait (Heterozygotes): 2 bands (HbS moves slower than HbA)
Sickle cell disease (Homozygotes): 10-30% HbF Beta-thalassemia major (β+/β+or β°/β°): 90% HbF (ɑ2ɣ2) Beta Thalassemia minor or trait (β+/β or β0/β): 3.6-8% HbA2 (ɑ2δ2); 5% HbF (ɑ2ɣ2) Alpha thalassemia:
|
|
| HbF determination | Qualitative estimation: Apt or Alkali denaturation test
Quantitative estimation: Kleihauer test (Acid elution method) |
|
| CD55/59 flow cytometry, Ham’s acidified serum test, Sucrose lysis test | Positive in Paraoxysmal Nocturnal Hemoglobinuria (PNH) | Negative |
| Causes | Complement mediated: Cold agglutinins, PNH, Incompatible red cell transfusions
Injury: Fibrin in vessel lumen: DIC, Vasculitis Physical trauma: Prosthetic valves, Small vessels of feet during hard marching (March hemoglobinuria) Chemical or thermal damage: Alpha toxin of Cl.perfringes, burns, snake venom, drugs in patient with G6PD deficiency |
Membrane defects: Hereditary spherocytosis, Hereditary elliptocytosis
RBC enzyme defects: G6PD deficiency, Pyruvate kinase deficiency Environmental: Infections, Drug-induced, Immune mediated, HUS Abnormal hemoglobins: Hemoglobinopathies, unstable hemoglobin |
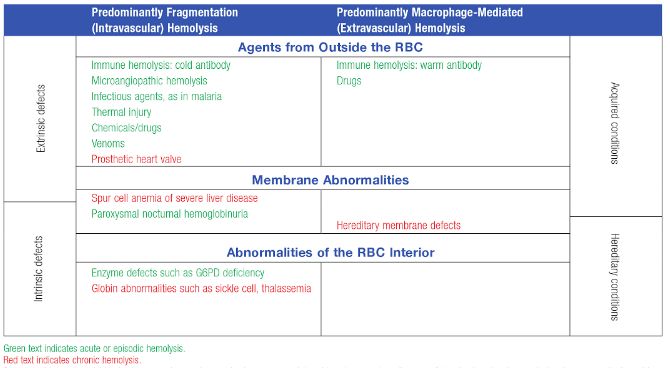
Acute vs Chronic Hemolysis
Acute hemolysis
- Rapid onset and is isolated (sudden), episodic or paroxysmal
- Normal between episodes
- Intravascular volume is intact and the clinical signs and symptoms are directly related to anemia.
- Fever and chills, acute chest and back pain, acute renal failure
- Example: Intravascular hemolysis in Paroxysmal nocturnal hemoglobinuria, Paroxysmal cold hemoglobinuria, ABO incompatibile blood transfusion
Compensated Chronic hemolysis
- RBC life span is chronically shortened, but bone marrow compensation prevents anemia.
- Dramatic acute hemolytic event occurs when the cells are challenged with oxidizing agents.
- No features of chronic hemolysis like gall stones and splenomegaly.
- Example: G6PD deficiency
Uncompensated Chronic hemolysis
- Chronic onset with extramedullary hematopoiesis inadequate to compensate severe anemia.
- Pigment gall stones
- Splenomegaly
- Example: Extravascular hemolysis in RBC membrane defects, Thalassemia, etc.
Inheritance of Hemolytic Anemia
Disorder of RBC membrane cytoskeleton (structural): Autosomal Dominant, .e.g. Hereditary spherocytosis
Disorder of globin chain (transport protein): Autosomal Recessive, e.g. Sickle cell anemia, Thalassemia
Disorder of RBC enzymes: Autosomal Recessive, e.g. G6PD deficiency, Pyruvate kinase deficiency
Only Acquired Intrinsic RBC defect: Paroxysmal Nocturnal Hemoglobinuria (PNH)
Most Extrinsic causes of hemolysis: Acquired
Defects in Specific Cause of Hemolytic Anemia
Hereditary Spherocytosis: Spectrin binds to Ion transporters in cell membrane Band 3 in head region and Glycophorin A in tail region with the help of Band 4.2 + Ankyrin and Band 4.1 + Actin respectively. Mutation in these proteins lead to Hereditary spherocytosis.
- Common mutations: Ankyrin (head region) > Band 3
- Uncommon mutations:Band 4.2 (Palladin), Spectrin, Glycophorin A
Hereditary Elliptocytosis: Commonset cause is mutation in spectrin.
G6PD deficiency: G6PD is required to produce NADPH which inturn is needed to produce Reduced Glutathione (GSH) required to convert hydrogen peroxide to water. Hence, oxidative stress (free radical release by leukocytes in infection, fava beans, antimalarials, sulfonamides, nitrofurantoin, etc.) in GSH deficient state leads to hemolysis. Also, denatured globin forms membrane-bound precipitaes known as Heinz bodies capable of inducing both intravascular and extravascular hemolysis. Episodes are self-limited because only old RBCs are lysed.
Paraoxysmal Nocturnal Hemoglobinuria (PNH): Bone marrow stem cells acquire mutations in Phosphatidyl Inositol Glycan A (PIGA) gene which is essential for synthesis of Glycosylphosphatidyl Inositol (GPI) anchor responsible for providing cell membrane attachment to complement inactivating proteins like:
- CD59 (Membrane inhibitor of reactive lysis)
- CD55 (Decay Accelerating Factor)
- C8 binding protein.
Acidotic states like exercise (increased lactic acid) or sleep (decreased respiratory rate and carbondioxide washout) activates complement system.
- Sleep at night = Nocturnal hemolysis
- Morning = Hemoglobinuria
Cold Agglutinin Autoimmune Hemolytic Anemia (AIHA): IgM directed against I antigen present on RBCs can:
- Agglutinate RBCs due to large size of IgM (pentamer)
- Activate complement resulting in opsonization and extravascular hemolysis of RBCs.
Cold Hemolysin AIHA: IgG (Donath-Landsteiner antibodies) directed against P antigen present on RBCs lead to intravascular hemolysis.
Sickle cell anemia: Point mutation that causes the glutamic acid to be replaced by valine at the beta-6 position of the globin chain.
- Deoxygenation = HbS becomes insoluble = Aggregation and polymerization = Sickling
- Increased HbS concentration = Increased sickling i.e. more severe in homozygous disease. HbA (in heterozygotes), HbC (lysine replaces glutamate at beta-6 position of globin chain) and HbF (newborn till 6 months and hydroxyurea) have inhibitory effect on disease.
- Increased MCHC = Increased sickling i.e. intracellular dehydration increases MCHC and sickling; thalassemia decreases MCHC and sickling.
- Acidosis shifts the Oxygen-Hemoglobin dissociation curve to right, increasing deoxygenated hemoglobin and hence, sickling.
- Sluggish circulation = increased duration of exposure to decreased oxygen tension = Increased sickling
- Initial reversible sickling: Increased adhesivenss in microcirculation = Vasocclusive crisis or Pain crisis (common) i.e. Dactylitis (Hand foot syndrome), AVN femoral head, Fish mouth appearance of vertebra, Extramedullary hematopoiesis, Acute chest syndrome, Stroke or seizure, leg ulcers, priapism, congestive splenomegaly (gamma gandy bodies)
- Repeated attacks cause irreversible sickling and oxidative damage = Splenic sequestration and extravascular hemolysis (most dangerous) = Hyperdynamic circulation and cardiomegaly
- Progressive infarction of spleen = Autosplenectomy
- Parvovirus attack = Aplastic crisis
Beta-thalassemia: Mutation (most common – splicing mutation) in chromosome 11 results in defective beta-globin synthesis.
- Defective beta-globin synthesis (decreased HbA1) but normal alpha-globin snythesis that increasingly combines with gamma-globin (increased HbF) and delta-globin (increased HbA2)
- Ineffective erythropoiesis, Extravascular hemolysis, Repeated blood transfusion = Iron overload and Secondary hemochromatosis (heart, liver, spleen, pituitary, hypothalamus, islets of langerhans) = Cardiac failure and Endocrine deficiency
- Anemia leading to hypoxia = Increased erythropoietin secretion = Increased extramedullary hematopoiesis
- Beta-thalassemia minor or trait = Resistance to falciparum malaria
Alpha-thalassemia: Gene deletion in chromosome 16 results in defective alpha-globin synthesis.
- Alpha-globin synthesis defective but normal beta-globin and gamma-globin synthesis.
- Tetramer of beta-globin: HbH = Not able to release oxygen to tissue = Ineffective erythropoiesis and Extravascular hemolysis
- Tetramer of gamma-globin = Barts Hb = Not able to transport oxygen properly = Intrauterine fetal hypoxia and death