Dosage and Absorption
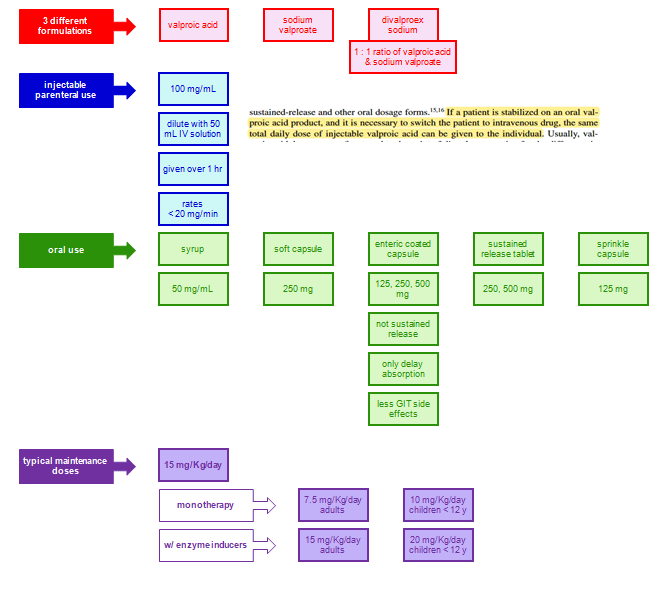
VPA is absorbed through the small intestine, and delayed- and extended-release forms are designed to extend the dosage interval and bypass the stomach to reduce dyspepsia.
The liquid and tablet forms have a more than 10-fold higher absorption rate constant than the extended-release forms and peak at 1 to 3 hours after ingestion.
The delayed-release divalproex sodium (which combines equal amounts of VPA and sodium valproate) has a lag time (T lag) of 2 hours and then releases quickly for small intestinal absorption at a rapid rate much like the liquid-filled capsule.
The extended-release form of divalproex slowly absorbs with time-to-peak of 8 to 16 hours (ka = 0.2) and provides for effcient 24 hours dosing in adolescents and adults.
For children, beyond the available liquid (ka = 4), tablet (ka = 4), and capsule (ka = 4) where three to four daily doses are needed to sustain adequate trough levels, the oversized sprinkle capsule (ka = 1.2, lag = 1 hour) provides sufficiently slow release to allow twice-daily dosing.
Complete bioavailability is evident for all the dosage forms except the extended release where 80% to 90% bioavailability is seen.
Co-ingested food moderately lengthens the max for the sprinkle capsule and can double it for the delayed-release tablet. Rectal administration of the syrup or suppositories can provide an alternative route to obtain therapeutic plasma levels.
Male and female adults differ in disposition for sustained-release valproate, because the concentration to dose ratio was observed as higher in adult females than in males.
This is likely less to do with differences in intrinsic hepatic clearance than the amount of enterohepatic recirculation, which is twice as high in females than in males.
This manifests in a larger area under the curve (AUC) for an equivalent dose and a significant “second peak” in females.
Mechanism of Action
Enhanced inhibitory neurotransmission by γ-amino butyric acid by VPA is done via increased synthesis, and decreased degradation and turnover. Additionally, anti-seizure effects derive from blockade of voltage-gated sodium channels and calcium channels.
Likely, the effects on serotonergic and dopaminergic receptor transmission modulate the benefits seen in migraine prophylaxis and the treatment of bipolar disease, depression, and other psychiatric and mood disorders.
The histone deacetylase inhibition observed with VPA is responsible for the encouraging experimental and clinical data seen with this drug against certain forms of cancer.
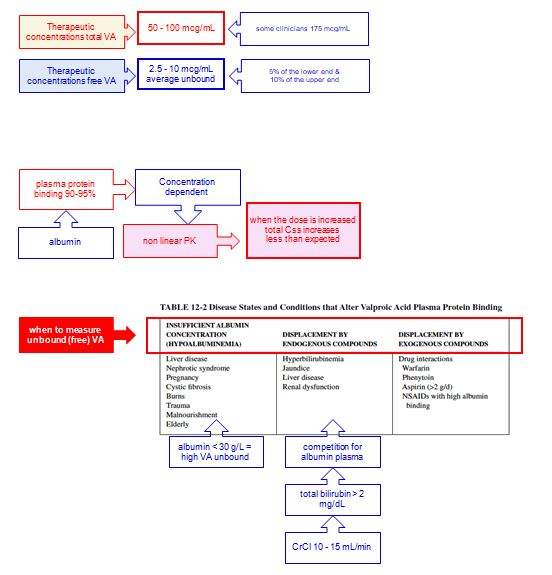
Therapeutic Concentration
Adverse effects
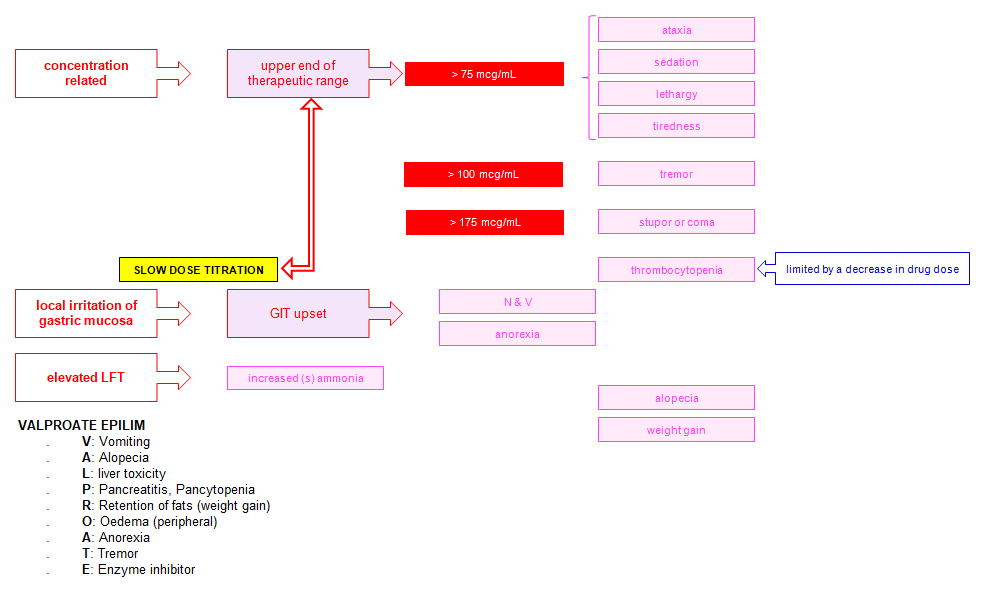
Mnemonic: VALPROATE
1. Vomiting
2. Alopecia
3. Liver toxicity
4. Pancreatitis, Pancytopenia
5. Retention of fat (weight gain)
6. Oedema (peripheral)
7. Anorexia
8. Tremor
9. Enzyme inhibitor
Metabolism and Clearance
METABOLISM AND CLEARANCE
Three main routes of metabolism produce about 10 to 20 metabolites for VPA, some purported to have CNS activity during long-term therapy. Glucuronidation to VPA-glucuronide is responsible for 40% to 50% of the disposition of VPA, predominantly using UGT A3 and UGT 2B7.
Mitochondrial β-oxidation accounts for 30% to 40%, and the cytochrome p450 system (mainly 2C9) a more minor 10% that produces hydroxylated metabolites and unsaturated moieties.
Children have more accentuated 2C9 activity compared to adults and utilize this pathway more, with longer time to full maturation of some glucuronidating enzymes.
This is one reason why the production of 4-ene VPA via this pathway, and its potential to cause fatal hepatotoxicity, is more selected toward younger children, with additional risks, including mitochondrial diseases, inborn errors of metabolism, and/or with polytherapy with enzyme-inducing antiepileptic drugs (AEDs).
Moreover, the therapeutic use of carnitine, in those at greater risk for hyperammonemia and drug-induced liver injury, in part prevents the incorporation of the toxic 4-ene VPA into mitochondria where 2,4-diene-VPA-CoA can be produced, itself a hepatotoxin.
Selected polymorphisms in UGT and 2C9 can reduce VPA clearance via these pathways,7 but phenotypic expression does not always follow genotype, because 2C9 expression can downregulate in epilepsy.43
Therefore, reduced function alleles do not consistently affect population-wide pharmacokinetics. Yet, normal 2C9 expressers consistently showed higher dose requirements than low expressers, show a higher VPA,43 and dose guidance using CYP2C9 genotype and phenotype expression in children more precisely predicted dose, achieved therapeutic range serum concentrations, and reduced hyperammonemia and other metabolic adverse effects.40
The same impact of 2C9 polymorphisms was not seen in adults, where no differences in concentration to dose ratio were observed between patients with wild-type and poor metabolism alleles.
Given the concentration-dependent protein binding and low extraction ratio (i.e., rate limitation is with intrinsic clearance), total VPA clearance increases with increasing unbound fraction:
CL hepatic = fu * CL intrinsic
fu = CL hepatic/CL intrinsic
Css = Dose * F/CL hepatic = (Dose * F)/(fu * CL intrinsic)
Therefore, as the fraction unbound increases with higher doses and total concentrations, so too the total clearance CL and total steady-state concentration Css will increase less-than-proportionately with increasing dose.
Because mathematically: C unbound = fu * C total
C total = C free/fu
The C total decrease and fraction unbound increase will offset each other, leaving a relatively stable unbound VPA concentration, but total levels must be interpreted carefully with recognition that the relationship between free levels and total levels changes when protein binding changes, and any factor altering intrinsic clearance, will alter the accumulation of free VPA concentrations.
Relatively reduced drug metabolic activity is observed in the elderly, with the concentration to dose ratio of VPA higher than that in young adults.
Decreased intrinsic clearance coupled with lower protein binding will often manifest as total drug concentrations being similar to younger adults but with higher free concentrations and clinical effect.
Furthermore, the greater likelihood of polytherapy and interacting medications in this age group makes the elderly of specific concern for VPA toxicity, with needs for closer clinical and therapeutic drug monitoring.
Clearances of VPA on monotherapy are within typical ranges of 12 to 25 mL/ hr/kg in children and 6 to 12 mL/kg/hr in adults with elimination half-lives ranging from 6 to 12 hours and 8 to 20 hours, respectively.
The half-life may be shorter when protein binding is lower, depending on whether expansion of Vd offsets the increase in total CL.
Larger clearance and shorter half-life are expected when enzyme-inducing co-medications are prescribed.
Increased intrinsic clearance of VPA is also recognized in traumatic brain injury.
[CP_CALCULATED_FIELDS id=”6″]
[CP_CALCULATED_FIELDS id=”7″]
[CP_CALCULATED_FIELDS id=”8″]
[CP_CALCULATED_FIELDS id=”9″]
[CP_CALCULATED_FIELDS id=”10″]
[CP_CALCULATED_FIELDS id=”11″]
[CP_CALCULATED_FIELDS id=”12″]
[CP_CALCULATED_FIELDS id=”13″]
[CP_CALCULATED_FIELDS id=”14″]
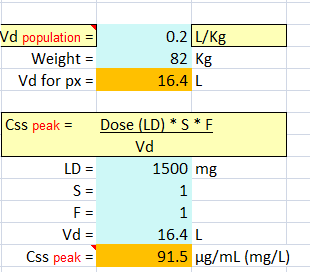
Question #1 B.I. is a 46-year-old, 82-kg man admitted to the neurointensive care unit after having serial tonic–clonic seizures emanating from a newly diagnosed glioblastoma.
Repeated bolus doses of levetiracetam were not useful in suppressing the seizure activity, and it is decided to administered VPA IV.
He has normal liver enzymes and function tests, and his albumin is 4.2 g/dL.
A 1,500 mg loading dose given over 30 minutes is prescribed, with a 30-minute postinfusion serum level to be obtained. What peak concentration might be expected?
Because the typical half-life of valproate in adults ranges between 10 and 20 hours, no significant drug loss would occur between the time to load dose and the time to peak concentration.
Therefore, the peak concentration can be estimated using the single-dose bolus model (Equation 15.1 ) assuming the average V or an adult is 0.2 L/kg.
Vd = 0.2 L/kg * (82 kg) = 16.4 L
C peak = (S) (F) (Loading dose)/Vd – 91.5 mg/L
| [CP_CALCULATED_FIELDS id=”15″] |
| [CP_CALCULATED_FIELDS id=”16″] |

Hard worker, Reliable, Team player, Family man.
MBChB (O’Porto Univ.), Dip HIV Man (CMSA), DTM&H (Wits), DipPEC (CMSA), Dip Internal Medicine (CMSA), M. Med. Clinical Pharm [Cum Laude] (Univ. Pretoria), FCP (CMSA)
What happens to blood level of valproate when taken with aspirin?