Table of Contents
Once again, I’m back with a biochemistry topic that everyone hates. Let’s try to break the topic, simplify it and cover all the important aspects of Glycogen Storage Diseases (GSD).
7 types of Glycogen Storage Diseases
Mnemonic:VP CAM HT. This is a pretty lame mnemonic for the order of the disease but this is what I’ve used for years now. If this doesn’t work for you, try this – Very Poor Carbohydrate Affects Muscle and Hepatic Target.
- Type I – Von Gierke’s disease
- Type II – Pompe’s disease
- Type III – Cori’s disease
- Type IV – Anderson’s disease
- Type V – McArdle’s disease
- Type VI – Her’s disease
- Type VII – Tauri’s disease
Enzymes affected by Glycogen Storage Diseases
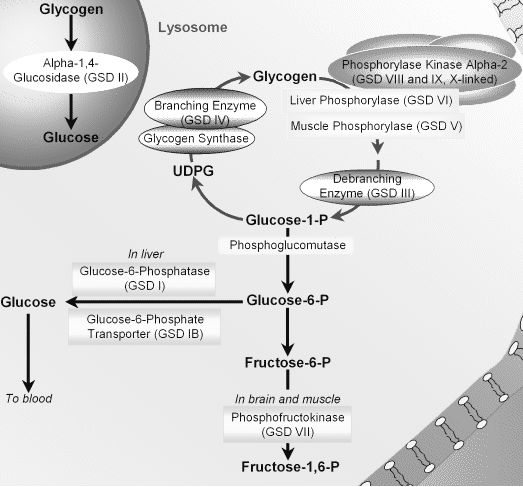
For Type III and Type IV Glycogen Storage Diseases:
Mnemonic: AB CD
- Anderson’s disease: Branching enzyme
- Cori’s disease: Debranching enzyme (Amylo 1,6 glucosidase)
For Type V and Type VI Glycogen Storage Diseases:
Mnemonic: M for Muscle and H for Hepatic.
- McArdle’s disease: Muscle glycogen phosphorylase
- Her’s disease: Hepatic glycogen phosphorylase
For remaining Type I, Type II and Type VII Glycogen Storage Diseases:
Mnemonic: GAP
- Von Gierke’s disease: Glucose-6-phosphatase
- Pompe’s disease: Acid maltase (Lysosomal alpha 1,4 – glucosidase)
- Tauri’s disease: Phosphofructokinase 1 (PFK-1)
Mode of Inheritance of Glycogen Storage Diseases
Inheritance of enzyme defects are by rule Autosomal Recessive. All are autosomal recessive diseases except Phosphorylase kinase deficiency (Glycogens storage disease type IXa, IXb) which is not mentioned above. Read Inheritance Pattern : Rule Mnemonics.
Problem in Glycogen Storage Disease
Children with Glycogen Storage Disease can make glycogen but cannot effectively catabolize it. Glycogen is thus stored in huge quantities in the liver. During periods of starvation, e.g. during an intercurrent viral illness, the children become hypoglycemic and lethargic.
Predominantly affected tissues in Glycogen Storage Disease
1. Hepatic forms: Type I, III, IV and VI
- Storage of glycogen in liver (hepatomegaly)
- Reduction of glucose in blood (hypoglycemia)
2. Myopathic forms: Type V and VII
- Glycogen deposition in muscle
- Muscle cramp after exercise
- Exercise induced lactic acidosis (block in glycolysis)
- There may be myoglobinemia
3. Type II (Pompe’s disease): Generalized form
- The enzyme affected is lysosomal alpha-1,4 glucosidase. The glycogen accumulates in lysosome and hence, there is generalized affection of organs.
- Pompe’s disease affects pump, i.e. heart. Infantile form is very severe and the infant dies with few months due to cardiac failure.
Clinical features of Glycogen Storage Disease
1. Clinically, GSD type I can be easily differentiated by:
- Massive hepatomegaly
- Metabolic features can be remembered in a simple way: there is defect of glucose 6 phosphatase –
- Glucose-6-phosphate cannot be converted to glucose between meals: Severe hypoglycemia (few hours after meal) not responsive to glucagons and adrenaline (metabolic block in formation of free glucose from liver glycogen).
- Glucose-6-phosphate accumulates:
- Undergoes Glycolysis: Generates pyruvate and then lactate; lactate cannot be recycled to gluconeogenesis in liver (lactic acidosis). Lactic acidosis inhibits urinary excretion of urates (hyperuricemia).
- Glucagon stimulation doesn’t improve hypoglycemia but rather leads to lactic acidosis.
- Undergoes HMP shunt: Generates excessive ribose-5-phosphate which generates purine nucleotides and they are degraded through the “salvage pathway” into urice acid (hyperuricemia)
- Increased glycolysis and decreased gluconeogenesis: Increased NADH, NADPH, glycerol and acetylCoA – hypertriglyceridemia.
- MalonylCoA derived from AcetylCoA inhibits carnitine acyltransferase: Beta-oxidation of fatty acid inhibited (no ketosis)
2. Hepatomegaly is seen in all except:
- Type 0 (Glycogen synthase deficiency)
- Type IV or Anderson’s disease (Hepatomegaly may be absent and death is early, i.e. in 2 years due to early cirrhosis)
Hepatomegaly with splenomegaly and other generalized signs of storage disorder, should suggest glycogen as the source of storage component causing hepatomegaly.
3. Myopathic forms (type V and VII):
- Muscle cramps and pain upon exertion and are easily fatigued.
- Otherwise life is normal (utilization of muscle glycogen is not essential for life).
- No increase in expected blood lactate after exercise (most important source of lactic acid during muscular activity is not free glucose from blood but stored muscle glycogen).
Investigations for Glycogen Storage Diseases
- Enzyme activity in blood leukocytes (all except type I GSD) and liver (type I GSD) and/or
- Molecular analysis of appropriate gene
Management of Glycogen Storage Diseases
Regular high carbohydrate meals during the day, and continuous feeds during the night (or uncooked cornstarch, a slow release form of glucose, every 4-6 hours).

He is the section editor of Orthopedics in Epomedicine. He searches for and share simpler ways to make complicated medical topics simple. He also loves writing poetry, listening and playing music.

Hey thanks man! Really helped memorizing and recalling storage disorders
Awesome summary and mnemonics!
Well thought out mnemonics. Keep up the good work!